Phylogenetic analyses
2019-09-11
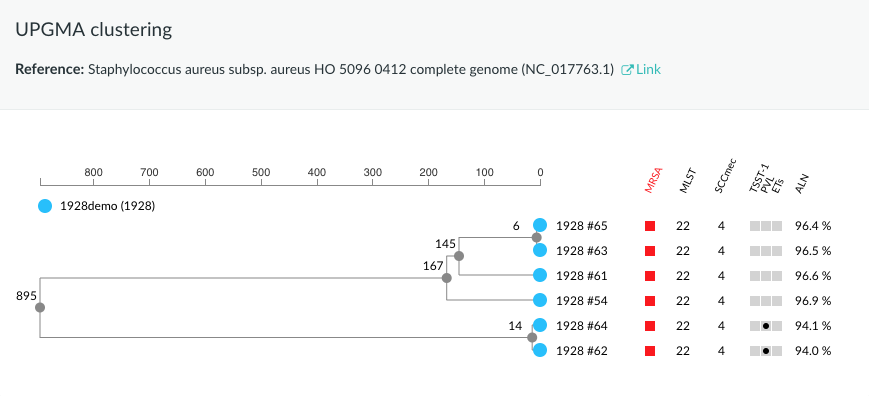
Samples originated from Harris SR et al., Whole-genome sequencing for analysis of an outbreak of meticillin-resistant Staphylococcus aureus: a descriptive study
On the 1928 platform, a phylogenetic tree of your uploaded Next-generation sequencing samples can be created by using two different methods, core genome multilocus sequence typing (cgMLST) and single nucleotide polymorphism (SNP) clustering. The clustering methods attempt to reconstruct phylogenies of samples and visualise them as dendrograms. Samples that are located close together on the dendrogram and have fewer counted differences are closely related. Samples that are too similar or identical on the dendrogram, may indicate a transmission event. Both cgMLST and SNP clustering methods use the UPGMA algorithm for the construction of dendrograms.
cgMLST
The cgMLST method identifies the alleles of the core genes of a sample. Core genes are genes that are found in most bacterial clades of a species. The sum of all core genes and their alleles for a species, comprise the species cgMLST schema. We have created a cgMLST schema for all supported species on the 1928 platform. The quality of the schemas has been verified by testing them against publicly available datasets.
When a sample is being analysed, part of the analysis is to determine the cgMLST profile of the sample. That is, to determine the alleles of the genes that are part of the species schema. The percentage of genes that have an allele assigned is being referred to as the “fraction of core”. The rest of the genes that haven’t got an allele assigned to them, are treated as genes that are missing from the sample’s genome. A cluster is deemed reliable if all samples are above 95% fraction of core, and samples bellow that value should be removed from that cluster to avoid confounding the phylogenetic tree.
A cgMLST cluster is being created by counting the number of different alleles in core genes, across samples and applying the UPGMA algorithm on the distance matrix of these samples. When counting the different alleles between two samples, we don’t include genes that are missing in the differences.
SNP
A SNP cluster is created by counting the number of nucleotides that differ across samples and applying the UPGMA algorithm on these differences. We perform a core-genome alignment, keeping only regions of genomes that have aligned across all samples. We count the nucleotides that differ on these regions; a position where the nucleotide is different across samples contains a SNP. A SNP cluster based on alignments in which all samples have aligned for a big percentage of their genomes is considered a good quality cluster. The more closely related the samples are, the better the alignment will be. This happens because closely related samples usually share larger portions of their genomes than more distantly related ones. Since we count SNPs on regions that have aligned on all samples that we cluster, the selection of samples will affect the cluster. Including just one divergent sample, would decrease the total portion of the genomes that are aligned between all samples.
Strengths and Weaknesses
By counting only different alleles between genes of samples, the strength of cgMLST clustering lies on providing enough resolution for the detection of outbreaks, without overinflating the amount of differences in a case where a single recombination event introduces many differences. In such an event, SNP clustering would portray the distance of a sample to the rest of the cluster in excess to the real phylogenetic relationship.
Additionally, the cgMLST clustering method doesn’t get affected from clustering distantly related samples, whereas the SNP clustering method is more reliable, the more closely related the samples are. For this reason, to get an overview of all samples, use cgMLST clustering. However, the SNP clustering method quantifies in greater detail the differences among genes and even intergenic regions between samples than cgMLST clustering. As such, use SNP clustering for zooming into a region of the phylogenetic tree of your samples.