Long-read sequences: What are they and how are they used?
2020-06-01
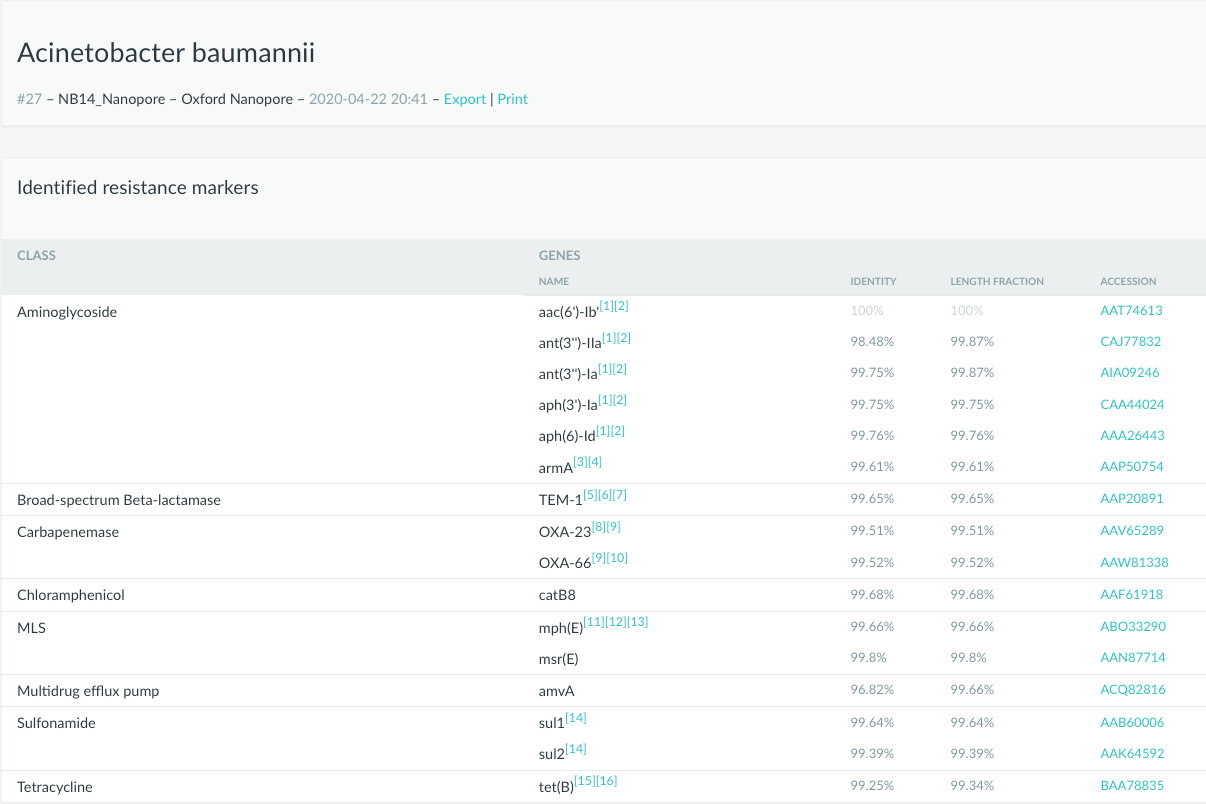
We are happy to inform you that our service now supports data generated from long-read sequencing.
A bacteria’s “fingerprint” is the unique sequence of tiny elements that together make up the bacteria’s genome.
Health care professionals collect samples from patients to see if any bacterial fingerprints are found in the patient. If there is a match between the bacteria’s genome and the genome found in the sample, the bacteria is identified in the patient.
Identifying genomes in a patient sample is not straight forward. Genomes are very long and current methods do not allow to extract a full genome sequence in one go. The sample genome is therefore broken down into smaller parts that are later assembled into a full genome that can be compared to the bacteria. These smaller parts are also called ‘reads’.
Assembling a genome from reads has an inherent risk of putting together the wrong sequences and hence obtaining a faulty result that can not match the patient sample’s genome with the bacteria’s genome. Therefore, the longer the reads are, the more accurate result is.
One method for sequencing a genome is called short-read sequencing which, as the name suggests, breaks up the sample genome into shorter parts that can later be assembled. This method is solid and widely used in labs.
A new method named long-read sequencing is a way of using longer reads from the sample. Using longer reads from the genome gives a more complete assembly because the reads are easier to distinguish and can therefore be assembled with a lower risk of faulty outcome.
Long-read sequencing is a new generation of sequencing and we are happy that our service now supports data generated via this sequencing method!